Web連携テキスト バイオインフォマティクス
最終更新: 2026/02/13
付録
R関連情報(第1章)
本書と直接関係のない部分もありますが、初心者の方は基本的にはざっとでよいので順番通りにやっていくとよいと思います。
R1.010 R本体とRStudioとパッケージのインストール
R1.020 RStudioの基本的な利用法
R1.030 1.2.1項の相補鎖など(基礎)
Rに慣れることを目的として、「1.2.1
サンガー法の原理」の冒頭部分で述べた相補鎖や逆相補鎖をRで得るやり方を解説します。まずはBiostringsパッケージをロードして、このパッケージが提供するcomplementやreverseComplement関数を使えるようにします。
library(Biostrings)
次に、本文中で示したものと同じ塩基配列をsequencという名前のオブジェクトに格納し、その中身を表示させます。背景が灰色になっている部分がコマンド、##から始まっている背景が白くなっている部分が実行結果です。当たり前といえば当たり前ですが、入力と同じ配列が出力されていることがわかります。
sequenc <- "AGTGACGGTCTT"
sequenc## [1] "AGTGACGGTCTT"
今取り扱っているsequencオブジェクトの中身は、見た目上明らかに塩基配列です。しかし、Biostringsパッケージが提供する相補鎖や逆相補鎖を得るための関数を利用するためには、これが塩基配列だと正しく認識させておかねばなりません。そこで次に、sequencを入力としてDNAStringSetという関数を実行し、その結果をfastaという名前のオブジェクトに格納する作業を行います。先ほどと同じく、背景が灰色になっている部分がコマンド、##から始まっている背景が白くなっている部分が実行結果です。背景が白の実行結果部分において、width列の12という数値が塩基数、seq列が実際の塩基配列になっていることがわかります。左下に見えている[1]はまだ気にしなくてもよいです。最初の行で見えているDNAStringSet objectというのが、(塩基)配列情報をBiostringsパッケージで取り扱うために定義された特有の形式なのだという理解でよいです。
fasta <- DNAStringSet(sequenc)
fasta## DNAStringSet object of length 1:
## width seq
## [1] 12 AGTGACGGTCTT
いよいよ本番1として、相補鎖を得るためのcomplement関数を実行します。入力はDNAStringSet形式のfastaオブジェクトです。確かに実行結果として見えているものは、本文中のものと同じ”TCACTGCCAGAA”です。
complement(fasta)## DNAStringSet object of length 1:
## width seq
## [1] 12 TCACTGCCAGAA
本番2として、逆相補鎖を得るためのreverseComplement関数を実行します。確かに実行結果として見えているものは、本文中のものと同じ”AAGACCGTCACT”です。なお、以下ではreverseComplement(fasta)としていますが、reverseComplement(x=fasta)と書いても構いません。
reverseComplement(fasta)## DNAStringSet object of length 1:
## width seq
## [1] 12 AAGACCGTCACTR1.040 エラーメッセージに慣れる
次に、存在しないオブジェクトugeの中身を見ようとして、どういうエラーが出るかを眺めます。このWebページはR
Markdownで自動的に作成しているため若干エラーメッセージの内容が異なっているかもしれませんが、「オブジェクト 'uge' がありません(object 'uge' not found)」のようなメッセージ表示されていることがわかります。このように、原因が明らかなエラーメッセージを眺めておけば、実際に遭遇する問題に対処しやすいです。
uge## Error in eval(expr, envir, enclos): オブジェクト 'uge' がありません
以下では、存在しないugeを入力として、逆相補鎖を返すreverseComplement関数の実行を試みています。重要なことは、エラーメッセージの中から対処すべき事柄が「オブジェクト 'uge' がありません(object 'uge' not found)」だけだと読み解くことです。
reverseComplement(uge)## Error in h(simpleError(msg, call)): 引数 'x' の評価中にエラーが起きました (関数 'reverseComplement' に対するメソッドの選択時): オブジェクト 'uge' がありません
逆相補鎖を返すreverseComplement関数は、入力としてDNAStringSet objectを受け付けます。そこで次に、ただの文字列であるsequencオブジェクトを入力として実行してみます。エラーメッセージだけを見ると、何をどうすればよいのかさっぱりわかりませんが、原因既知状態で眺めると穏やかに原因が特定できると思います。たとえば「..."character"...」が目につくと思いますが、sequenc <- "AGTGACGGTCTT"からも明らかなように、sequencの中身はcharacterです。
sequenc <- "AGTGACGGTCTT"
reverseComplement(sequenc)## Error: 関数 'reverseComplement' (シグネチャ 'x = "character"' に対する) に対する継承メソッドを見付けることができません
任意のオブジェクトを入力としてclass関数を実行すると、そのオブジェクトがどういうものかを調べることができます。class実行結果も、エラーへの対応(これをデバッグといいます)を行う上で有用な情報となります。
class(sequenc)## [1] "character"fasta <- DNAStringSet(sequenc)
class(fasta)## [1] "DNAStringSet"
## attr(,"package")
## [1] "Biostrings"
次に、存在しないugeオブジェクトを入力として、再度reverseComplement(uge)の実行結果を眺めます。よく見ると、自分が指定した覚えのない「引数
‘x’
の評価中にエラーが起きました」のようなエラーメッセージが出ていることがわかります。これは、この関数の場合は、入力であるDNAStringSet objectを受け付ける引数名がxだからです。それがわかっていれば、「reverseComplement(uge)で入力として与えたugeの引数名がxだな。なのでこれは引数名を省略しているが、実際にはreverseComplement(x=uge)だからこのようなメッセージが出ているのだろう」と解釈できるようになります。
reverseComplement(uge)## Error in h(simpleError(msg, call)): 引数 'x' の評価中にエラーが起きました (関数 'reverseComplement' に対するメソッドの選択時): オブジェクト 'uge' がありません
相補鎖や逆相補鎖を得る程度なら、入力として与える情報は1つだけです。しかし複雑な関数になってくると関数内で複数のオプション(引数)を与えて実行します。複数オプションがある場合は、その関数が想定するデフォルトの順番通りだと引数名を明示しなくても構いません。しかし、入力する側が理解しやすいように引数名を明示することもあります。このあたりのポリシーはヒトそれぞれですので、ユーザ側がこの段階で慣れておくとよいと思います。?reverseComplementは相補鎖を返すreverseComplement関数のマニュアルを表示するコマンドです。help(reverseComplement)でも同じ挙動になります。
?reverseComplement若干見栄えは異なりますが、上記実行結果とほぼ同じ情報を以下に示します。①や②で見えているxが、これらの関数で入力として与える情報の引数名です。RStudio上で上記コマンドを実行すると、RStudio画面右下のHelpタブで小さく表示されて見づらい場合は、たとえばBiostringsパッケージのページからリンクが張られているReference
Manualで、調べたい関数名でページ内検索する手もあります。以下の図は「reverseComplement」でページ内検索してとったスクショです。

R1.050 1.2.1項の相補鎖など(応用)
RStudio上で塩基配列を手入力し、実行結果がRStudio上に表示されるだけだと実感がわかない方もいらっしゃると思います。ここでは、塩基配列(AGTGACGGTCTT)ををFASTA形式ファイルとして独立に用意したもの(R1.050.fasta)を入力として用いるやり方を示します。以下は、入力ファイルとしてR1.050.fastaを指定して、Biostringsパッケージをロードして、このパッケージが提供するreadDNAStringSet関数を用いて読み込んだ結果をfastaという名前のオブジェクトに格納し、表示させるところまでのスクリプトです。背景が灰色になっている部分がコマンド、##から始まっている背景が白くなっている部分が実行結果です。背景が白の実行結果部分において、width列の12という数値が塩基数、seq列が実際の塩基配列、そしてnames列の情報がFASTA形式ファイルのdescription行部分に相当することがわかります。
in_f <- "R1.050.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta## DNAStringSet object of length 1:
## width seq names
## [1] 12 AGTGACGGTCTT hairetsu
次に、逆相補鎖を得たのち、それをout_fで指定した出力ファイルに保存するまでのスクリプトを示します。reverseComplement(fasta)実行結果をoutオブジェクトに格納し、それをwriteXStringSet関数を用いて保存していることが読み解けます。writeXStringSet関数実行時に、最初の引数としてoutを指定しているのは納得できると思います(逆相補鎖情報を含むoutオブジェクトの中身を保存したいからです)。次に出力ファイル名の引数に相当するfileという引数で、出力ファイル名情報が格納されているout_fを与えています。出力形式は、この場合は"fasta"形式の一択になりますが念のため明示しています。widthという引数では、1行あたり何塩基とするかを指定するオプションになります。この場合は12塩基しかないので特に気にする必要はありませんが、染色体など長い配列になると、width=60などを指定して1行あたりの塩基数を多めに設定するのを好むヒトもいます。
in_f <- "R1.050.fasta"
out_f <- "R1.050_out.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
out <- reverseComplement(fasta)
writeXStringSet(out, file=out_f, format="fasta", width=50)
念のため、以下に逆相補鎖情報を含むoutオブジェクトの中身を示しておきます。エラーなく実行できたヒトは、出力ファイル名として指定したR1.050_out.fastaが作業ディレクトリ上に保存されているはずです。
out## DNAStringSet object of length 1:
## width seq names
## [1] 12 AAGACCGTCACT hairetsuR1.060 1.2.2項のクオリティスコア
(1.1)式は、ある塩基のベースコールがエラーとなる確率pを入力として、その塩基のクオリティスコアqを返すものです。本文中でいくつかの入力と出力の関係をいくつか例示していますが、簡単ですのでRStudioでの計算例を示します。以下は、p = 0.01の結果です。確かに出力結果としてq = 20が得られていることがわかります。
p <- 0.01
q <- -10*log(p, base=10)
q## [1] 20
次は、数値ベクトルを入力として与えるやり方を示します。たとえば、p =
0.0001のときのqは40だと読み解きます。
p <- c(0.1, 0.01, 0.001, 0.0001)
q <- -10*log(p, base=10)
q## [1] 10 20 30 40
この場合のqは、4つの要素からなる数値ベクトルです。ベクトルの要素数を調べるlength関数の入力として与えて実行すると、確かに4という結果が返されます。
length(q)## [1] 4
ベクトルの2番目の要素情報を取り出したい場合はq[2]、4番目の要素情報を取り出したい場合はq[4]のように指定します。
q[2]## [1] 20q[4]## [1] 40
このベクトルqの5番目の要素は存在しないことが分かっていますが、もしq[5]とやると、Not
Availableを意味するNAという結果が返されます。
q[5]## [1] NAR1.070 1.7.1項のk-mer解析
1.7.1項の終盤から1.7.2項の最初のほうで述べているk-mer解析に関する基礎的な事柄に加えて、ショートリードデータを用いたゲノムサイズ推定までの内容を参考資料として以下に提供します。特に、ゲノムサイズ推定は、1.8.2項でも述べたものです。元ネタは、2022年5月17日に行った計3時間分の講義資料です。講義で利用されたい方は、pptx版もありますので、必要に応じてご自由に改変してご利用ください。
R1.080 1.8.4項の例題1.1(基礎)
1.8.4項の最後にある例題1.1をRでやることもできます。この項目の主目的は、ちょっとややこしいですがそれができるのを実感することです。readDNAStringSet,
strsplit, nchar, rep,
cなどのRの関数や、Rのノリに慣れてもらうことも兼ねています。ここでは、図1.8aの塩基配列をFASTA形式ファイルとして独立に用意したもの(R1.080.fasta)を入力として用いるやり方を示します。以下は、入力ファイルとしてR1.080.fastaを指定して、Biostringsパッケージをロードして、このパッケージが提供するreadDNAStringSet関数を用いて読み込んだ結果をfastaという名前のオブジェクトに格納するところまでのスクリプトです。
in_f <- "R1.080.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
以下では、入力ファイルの中身に相当するfastaオブジェクトを表示させています。背景が灰色になっている部分がコマンド、##から始まっている背景が白くなっている部分が実行結果です。DNAStringSetという独特の形式でFASTA形式ファイルの中身が格納されているのだと理解すればよいです。width列の60という数値が塩基数、seq列が実際の塩基配列の最初と最後の部分、そしてnames列の情報がFASTA形式のdescription行部分に相当します。随分冗長な表現方法だと思われるかもしれませんが、たとえば入力が数十個の染色体配列から構成されるヒトゲノム配列ファイルを読み込んだ場合をイメージしていただければ納得しやすいかと思います。
fasta## DNAStringSet object of length 1:
## width seq names
## [1] 60 TTGACAATTTCCTGATTCGGACG...ATGAATCCCTCAACTCTATTGCA fig1.8a
以下では、仮想制限酵素の認識配列がACなので、それを用いてfastaオブジェクト内の塩基配列(文字列なのでstring)を分割(split)するstrsplit関数を実行しています。実行結果をhogeオブジェクトに格納しています。2行目のstrsplit関数実行部分の右側に[[1]]がついていますが、簡単にいうと話をややこしくしないようにするためのおまじないです。なお、strsplit関数実行部分で「Warning in .normarg_at2(at, x): 'at' names were ignored」という警告メッセージが表示されていますが、エラーではないので気にする必要はありません。
補足:例題1.1をRでやるのは、あくまでもおまけ程度の位置づけです。例題番号的にこの位置にしましたが、結果的にちょっとややこしい内容のものが含まれています。それゆえ、初心者の方は「Rだとこんな感じでできる」ことを実感する程度の位置づけでよいと思います。もう少し真面目に解説すると、strsplit関数の出力がリスト形式とよばれるもので、そこから情報を取り出す際に大括弧(角括弧)が2つ並んだ[[1]]のような感じで情報を取り出しています。この場合は配列が1つしかないので、[[1]]として1番目の要素を取り出しています。
param <- "AC"
hoge <- strsplit(fasta, param)[[1]]## Warning in .normarg_at2(at, x): 'at' names were ignored
以下では、hogeの中身を表示しています。hogeの中身と例題1.1の解答例を見比べればわかりますが、ACで分割後の配列数(=5)が一致していることがわかります。また、2番目以降の配列の冒頭にACが付加されていないことがわかりますが、これはstrsplit関数の性質上やむを得ません。もしかしたら制限酵素認識配列専用の関数を誰かが作っているのかもしれませんが、実用上は自分が知っている関数の中から近い機能を果たすものをうまく使い、残りはad
hoc(その場しのぎ、のようなイメージでよいです)に対応するという感じでよいと思います。
hoge## DNAStringSet object of length 5:
## width seq
## [1] 3 TTG
## [2] 15 AATTTCCTGATTCGG
## [3] 5 GTCCC
## [4] 20 TATATCATATGAATCCCTCA
## [5] 9 TCTATTGCA
ad
hocな対応の例として、たとえばwidth(hoge)とやることで、hogeオブジェクト中の計5配列の長さ情報を数値ベクトルとして得ることができます。
width(hoge)## [1] 3 15 5 20 9
また、今は仮想制限酵素が2塩基だとわかっていますので、以下のようにwidth(hoge) + 2とすることで、ベクトル中の全要素に2を足すことができます。
width(hoge) + 2## [1] 5 17 7 22 11
上記は、paramオブジェクトの中身が2文字から構成されることが分かっているのでそのまま2としました。もちろんもう少し一般性を高めた表現も可能です。例えば、オブジェクト中の文字数をカウントするnchar関数を用いて、nchar(param)として利用することもできます。こんな感じで少しずつRの関数に慣れていくとよいでしょう。
nchar(param)## [1] 2width(hoge) + nchar(param)## [1] 5 17 7 22 11
ただ上記の結果だと、最初の要素にまで2が足されてしまっています。それゆえ、何を足すかという情報をあらかじめ作成しておいてから、width(hoge)の対応する要素と足し合わせるようなことをすればよいです。この場合は、1番目が0、2-5番目の要素が2の計5つの要素からなる数値ベクトルを作成して足すようなイメージです。rep関数は、1番目の引数として指定した情報を、2番目の引数で指定した個数だけ複製(replicate)します。以下では、rep(2, 4)として2を4つ作成しています。引数名を明示すると、rep(x=2, times=4)と書くこともできます。
rep(2, 4)## [1] 2 2 2 2
以下は、rep(nchar(param), 4)として、実質的に中身が2のnchar(param)にしているだけです。どちらでもいいじゃないかと思われるかもしれませんが、こんな感じで一般式で書くことによって手作業で変更せねばならないところを極力減らして汎用性を高めていきます。
rep(nchar(param), 4)## [1] 2 2 2 2
以下では、ベクトルを連結(combine)するc関数を用いて、最初の要素が0、2番目以降が2という数値ベクトルを連結させて得たものをtasuyouというオブジェクトに格納しています。ここまでで、width(hoge)の対応する要素に足すべき数値が格納されたtasuyouオブジェクトの完成です。Rの教え方はヒトそれぞれですが、このあたりを見るだけでも基本テクの習得が重要であることがわかると思います。
tasuyou <- c(0, rep(nchar(param), 4))
tasuyou## [1] 0 2 2 2 2
残りは、width(hoge)とtasuyouの対応する要素を以下のように足せばよいです。ここまでで例題1.1の解答例の最後に示した光学マップ情報が得られたことになります。
width(hoge) + tasuyou## [1] 3 17 7 22 11R1.090 1.8.4項の例題1.1(応用)
上記項目の続きですが、RStudio起動直後の状態でも、以下を実行すれば必要な情報を取得できます。この項目の主目的は、replaceAt関数の紹介や、as.character関数実行結果オブジェクトの取り扱いに関する小技の伝授です。なお、strsplit関数実行部分で「Warning in .normarg_at2(at, x): 'at' names were ignored」という警告メッセージが表示されていますが、エラーではないので気にする必要はありません。
in_f <- "R1.080.fasta"
param <- "AC"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
hoge <- strsplit(fasta, param)[[1]]## Warning in .normarg_at2(at, x): 'at' names were ignored
ACで分割された配列からなるhogeオブジェクト中の各配列に対して、1番目の塩基の位置にparamで指定した塩基(つまりAC)を挿入することもできます。これも本当は2番目以降の配列のみに挿入したいのですが、ややこしくなるのでreplaceAtという関数を用いればこういうこともできるんだという程度の情報にとどめておきます。以下では、param,
hoge,
replaceAt(hoge, 1, value=param)の実行結果を一気に表示しています。背景が灰色になっている部分がコマンド、##から始まっている背景が白くなっている部分が実行結果です。
param## [1] "AC"hoge## DNAStringSet object of length 5:
## width seq
## [1] 3 TTG
## [2] 15 AATTTCCTGATTCGG
## [3] 5 GTCCC
## [4] 20 TATATCATATGAATCCCTCA
## [5] 9 TCTATTGCAreplaceAt(hoge, 1, value=param)## DNAStringSet object of length 5:
## width seq
## [1] 5 ACTTG
## [2] 17 ACAATTTCCTGATTCGG
## [3] 7 ACGTCCC
## [4] 22 ACTATATCATATGAATCCCTCA
## [5] 11 ACTCTATTGCA
strsplit関数の入力は、入力が塩基配列であることを明示したDNAStringSet形式のfastaオブジェクトではなく、ただの文字列(as
character)としたものを与えることもできます。この場合は、as.character(fasta)を入力とすればよいです。
fasta## DNAStringSet object of length 1:
## width seq names
## [1] 60 TTGACAATTTCCTGATTCGGACG...ATGAATCCCTCAACTCTATTGCA fig1.8aas.character(fasta)## fig1.8a
## "TTGACAATTTCCTGATTCGGACGTCCCACTATATCATATGAATCCCTCAACTCTATTGCA"
以下を実行して得られる、出力結果のhoge2を眺めると、$fig1.8aというのが最初のほうに見えていて気持ち悪いと思われるかもしれません。慣れてくると、こういうの見た段階でhoge2$fig1.8aのような感じで取り扱えばよいと閃くことができるようになります。
hoge2 <- strsplit(as.character(fasta), param)
hoge2## $fig1.8a
## [1] "TTG" "AATTTCCTGATTCGG" "GTCCC"
## [4] "TATATCATATGAATCCCTCA" "TCTATTGCA"
たとえば、hoge2で見えている計5配列の配列長を調べたい場合は、hoge2$fig1.8aを入力として、文字数をカウントするnchar関数を実行すればよいです。
nchar(hoge2$fig1.8a)## [1] 3 15 5 20 9また、見慣れたDNAStringSet形式にしたい場合は、hoge2$fig1.8aを入力として、DNAStringSet関数を実行すればよいです。
DNAStringSet(hoge2$fig1.8a)## DNAStringSet object of length 5:
## width seq
## [1] 3 TTG
## [2] 15 AATTTCCTGATTCGG
## [3] 5 GTCCC
## [4] 20 TATATCATATGAATCCCTCA
## [5] 9 TCTATTGCAR1.100 1.10.2項のGFF3(ファイル取得)
1.10.2項で述べた、アノテーション結果を格納する代表的な形式であるGFF3ファイルをRStudioで読み込んで取り扱うやり方を示します。ここではまず、Ensembl (Cunningham et al., Nucleic Acids Res., 2022)のバクテリア版であるEnsemblBacteria (Release 54)上で、乳酸菌の1つであるLactobacillus casei 21/1のページを訪れて、全体像を把握します。以下はそのスクショです。まずは、このゲノムの基本情報を把握すべく、①または②のどちらでもよいのでクリックします。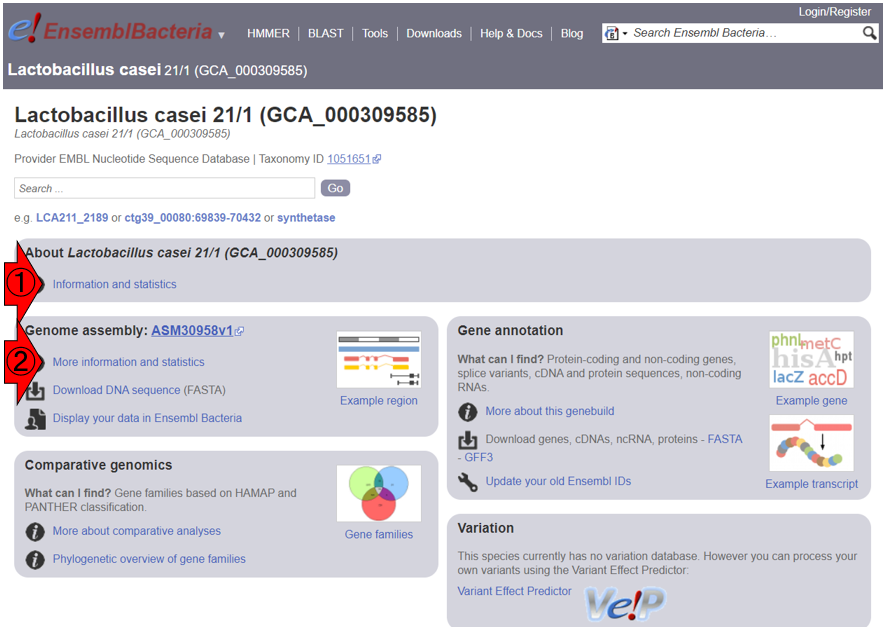
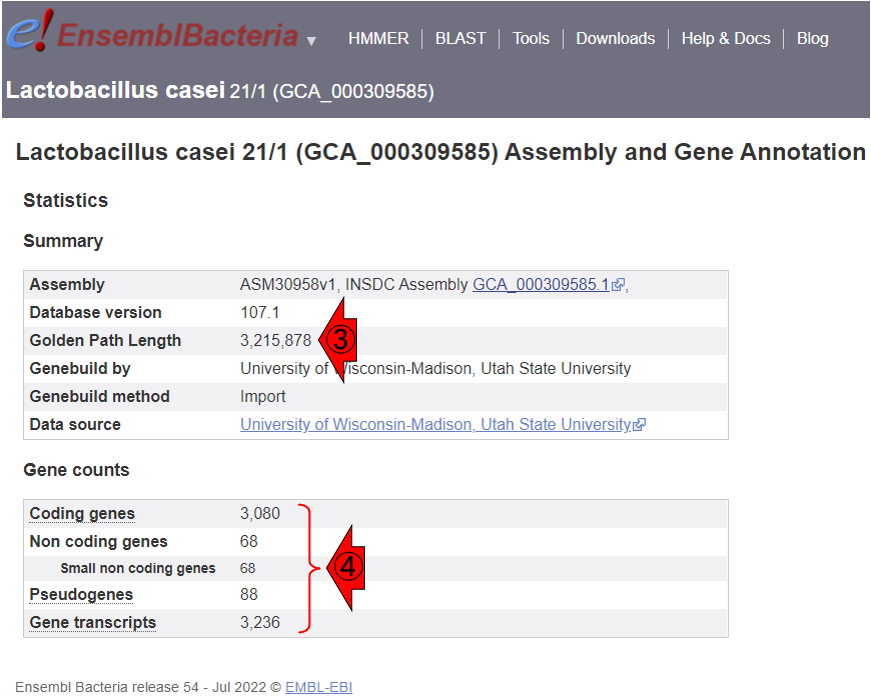
③より、このゲノムサイズは3,215,878 bp (約3.2 MB)だということがわかります。また④より、coding genesが3,080個、non-coding genesが68個、pseudogenesが88個、転写物全体としては3,080 + 68 + 88 = 3,236個だということがわかります。
次に、一旦前のページに戻ります。GFF3ファイルは⑤から取得可能です。ゲノム配列ファイルは⑥から取得できますが、⑦のFASTA形式ファイルの中からも取得できます。なお、ゲノム配列ファイルについては、1.10.3項で述べる反復配列をマスクしないバージョン(‘dna’)とマスクしたバージョン(マスクの程度によってさらに’dna_rm’と’dna_sm’があります)など様々なものが用意されています。ここではマスクしないバージョンのものを取り扱います。なお、ファイルはすべてgzipという形式(拡張子は.gz)で圧縮されていますので、圧縮・解凍ソフトで解凍せねばなりません(本当はFASTA形式ファイルは圧縮ファイルでも
readDNAStringSet関数で読み込めますが、gff3のほうがダメなので全部解凍しといたほうがややこしくないと思います)。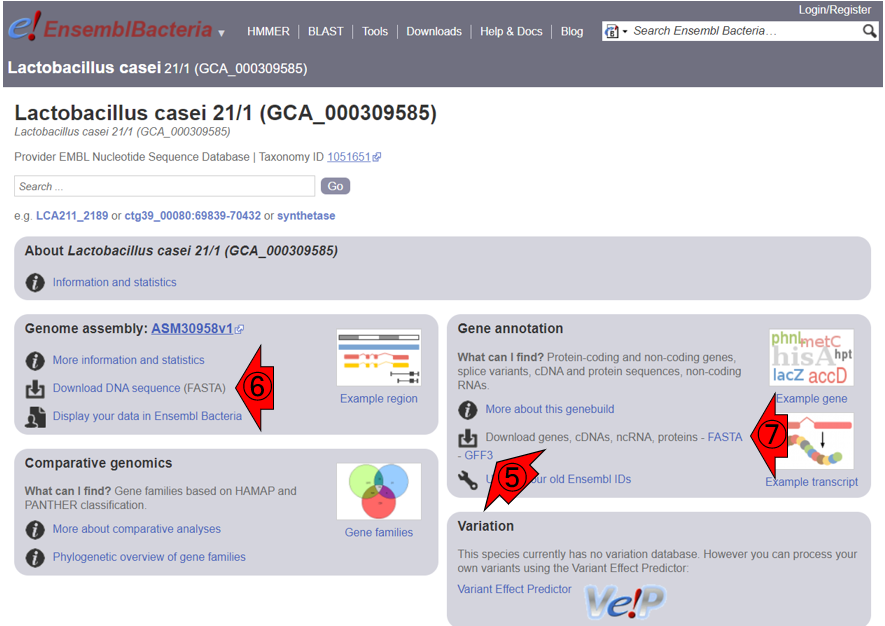
上記の情報はEnsemblBacteria (Release 54)のものですが、リリースの違いによってリンク切れになったりします。それゆえ、以降の解析が数年後でも可能となるよう、念のため以下でも解凍後のファイルへのリンクを貼っておきます:
- Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.49.gff3
- Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.cdna.all.fa
全部で3,168配列あります。3,080 coding genes + 88 pseudogenesと一致しています。
- Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.cds.all.fa
全部で3,080配列あります。3,080 coding genesと一致しています。
- Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.dna.toplevel.fa
全部で75配列あります。総塩基数は3,215,878 bpですので、確かにゲノムサイズと一致しています。
- Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.ncrna.fa
68配列あります。non-coding genesと一致しています。
- Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.pep.all.fa
全部で3,080配列あります。3,080 coding genesと一致しています。ただしこれはアミノ酸配列なので、読み込み時に用いる関数はreadDNAStringSetではなくreadAAStringSetにせねばなりません。
R1.110 1.10.2項のGFF3(読込)
1.10.2項で述べた、アノテーション結果を格納する代表的な形式であるGFF3ファイルをRStudioで読み込むやり方を示します。rtracklayerパッケージ(Lawrence et al.,
Bioinformatics, 2009)が提供するreadGFF関数や、GenomicsFeatures(Lawrence et al., PLoS
Comput Biol.,
2013)が提供するmakeTxDbFromGFF関数がありますが、ここでは後者のやり方を示します。GenomicsFeaturesを未インストールの方は、まず以下を実行してください。
BiocManager::install("GenomicFeatures", update=F)
次に、GenomicsFeatures(Lawrence et al., PLoS
Comput Biol.,
2013)が提供するmakeTxDbFromGFF関数を用いて、Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.49.gff3を入力として読み込むやり方を示します。背景が灰色になっている計4行から構成される以下のコマンドでは、まず入力ファイルをin_fというオブジェクト名で取り扱うという宣言をしています。次に、GenomicsFeaturesパッケージをlibrary関数を用いてロードしています。これでGenomicFeaturesが提供する関数を利用可能になったので、(入力ファイル名に相当する)in_fを入力としてmakeTxDbFromGFF関数を実行した結果をtxdbというオブジェクトに格納し、その中身を表示しています。
in_f <- "Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.49.gff3"
library(GenomicFeatures)
txdb <- makeTxDbFromGFF(in_f, format="auto")## Error in load_package_gracefully("txdbmaker", "starting with BioC 3.19, ", : Could not load package txdbmaker. Is it installed?
##
## Note that starting with BioC 3.19, calling makeTxDbFromGFF() requires
## the txdbmaker package. Please install it with:
##
## BiocManager::install("txdbmaker")txdb## Error in eval(expr, envir, enclos): オブジェクト 'txdb' がありません
背景が白の実行結果の冒頭部分にTxDb objectと書かれていることと、makeTxDbFromGFF関数を実行した結果であることから想像できるように、makeTxDbFromGFF関数はGFF形式ファイルを読み込んでTxDbというGenomicsFeaturesパッケージ内で有効な独特の形式でGFF情報を格納しているのだと理解すればよいです。重要なことは、無事に読み込めることと、txdbの中身に相当する背景が白の実行結果から見られる情報として「Nb of transcripts: 3236」に注目することです。この3236という数値は、確かにダウンロード元であるLactobacillus
casei 21/1から辿れるMore
information and statisticsで見られるGene
transcriptsの数と一致しています。GFFファイル自体は1つしかないため、すべての情報を含んでないと都合が悪いのだと考えれば納得できると思います。念のために以下にもリンク先のスクショを示しておきます。①の部分の数値と一致しているということです。
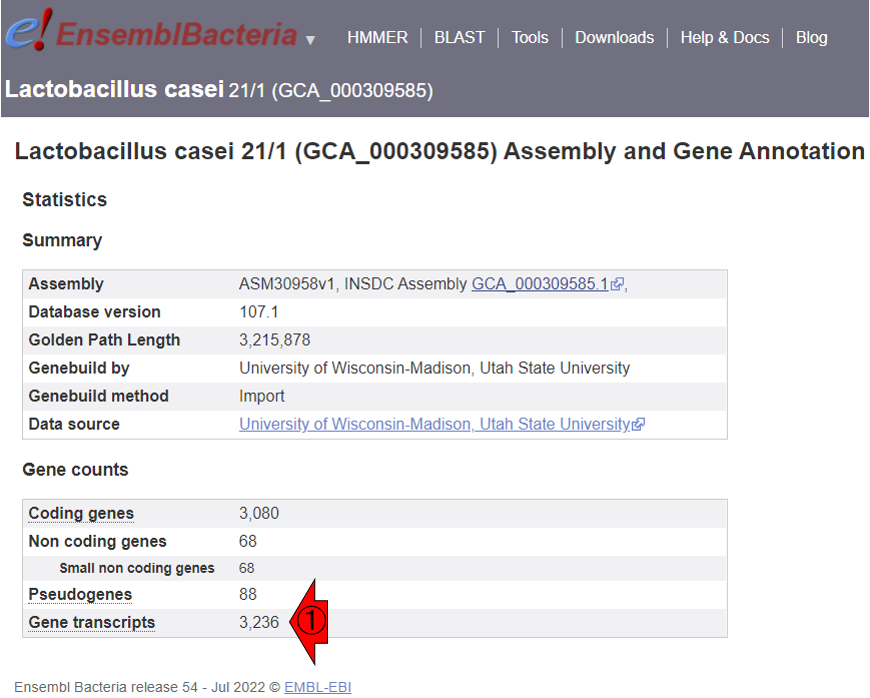
R1.120 1.10.2項のGFF3(座標取得)
TxDb objectであるtxdbオブジェクトだけを見せられても実感がわかないと思います。txdbの中に含まれているのは、ゲノム配列中のどこにどのようなフィーチャ(アノテーションがつけられるゲノム上の各領域のこと)があるのかという座標情報です。たとえばtxdbを入力としてtranscripts関数を実行すると、GRanges objectという形式の、この場合はtranscriptsなので転写物の座標情報を得ることができます。
seqnames列は、アノテーションに用いた大元のゲノム配列ファイル(Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.dna.toplevel.fa)内のdescription行で見られる配列名情報に相当します。1番目の転写物はctg01_00128という配列上、3236番目の転写物はctg75_00141という配列上に存在するものだと解釈します。ctgは、コンティグ(contig)を意味します。最もよくつながっているのが染色体レベル、その前段階がスカッフォールドレベル、そしてコンティグレベルとなります。この乳酸菌ゲノムは全部で75配列から構成されますが、バクテリア程度のゲノムサイズで75配列に分断されている程度のアセンブリ結果しか得られていないので、ある程度業界歴が長くなると「確かにコンティグレベルだろうな」というのがわかってきます。
ranges列はstartとendの位置情報、strand列は+鎖か-鎖のどちらかの情報、tx_id列は転写物のシリアル番号、tx_name列は転写物名を表しています。
in_f <- "Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.49.gff3"
library(GenomicFeatures)
txdb <- makeTxDbFromGFF(in_f, format="auto")## Error in load_package_gracefully("txdbmaker", "starting with BioC 3.19, ", : Could not load package txdbmaker. Is it installed?
##
## Note that starting with BioC 3.19, calling makeTxDbFromGFF() requires
## the txdbmaker package. Please install it with:
##
## BiocManager::install("txdbmaker")transcripts(txdb)## Error in h(simpleError(msg, call)): 引数 'x' の評価中にエラーが起きました (関数 'transcripts' に対するメソッドの選択時): オブジェクト 'txdb' がありません
ノリさえわかってくれば、txdbを入力としてpromoters関数を実行することでプロモータ配列を取得することもできます。以下は、転写開始点(TSS)の上流50塩基、下流10塩基の領域情報を取得するコマンドです。たとえば1番目の転写物のTSSは、ctg01_00128という配列の4769番目の塩基です(+鎖なので、start位置がTSSです)。この転写物のプロモータ領域は4719-4778となっていますが、確かにTSSである4769
- 50 = 4719、そして4769 + 10 - 1 =
4778で妥当な結果です。もう1つの例として、3236番目の転写物物は-鎖上にあるので、転写開始点(TSS)はctg75_00141という配列の639番目の塩基になります。それゆえ、上流50塩基は639
+ 50 = 689番目の塩基、そして下流10塩基は639 - 10 + 1 =
630番目の塩基になります。したがって、取得したプロモータ領域は妥当です。
in_f <- "Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.49.gff3"
library(GenomicFeatures)
txdb <- makeTxDbFromGFF(in_f, format="auto")## Error in load_package_gracefully("txdbmaker", "starting with BioC 3.19, ", : Could not load package txdbmaker. Is it installed?
##
## Note that starting with BioC 3.19, calling makeTxDbFromGFF() requires
## the txdbmaker package. Please install it with:
##
## BiocManager::install("txdbmaker")promoters(txdb, upstream=50, downstream=10)## Error in h(simpleError(msg, call)): 引数 'x' の評価中にエラーが起きました (関数 'promoters' に対するメソッドの選択時): オブジェクト 'txdb' がありませんR1.130 1.10.2項のGFF3(配列取得1)
ここでは、実際に取得した座標情報であるGRanges objectを利用して、その領域の塩基配列を取得します。ただし、GFF3ファイルの情報はただの座標情報にすぎないため、大元のGFF3ファイルと対応したゲノム配列も読み込んで利用せねばならない点に注意が必要です。ここではアノテーションに用いた大元のゲノム配列ファイル(Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.dna.toplevel.fa)も同時に読み込んでin_f1というオブジェクト名で利用しています。Rsamtoolsパッケージが提供するgetSeqやFaFile関数も利用します。Rsamtoolsを未インストールの方は、まず以下を実行してください。
BiocManager::install("Rsamtools", update=F)
以下は、アノテーションのGFF3形式ファイルとゲノム配列のFASTA形式ファイルの2つを入力として与えて、最終的にDNAStringSeq形式のfastaオブジェクトを得るところまでのコードです。計3つのパッケージを明示的にロードしていることがわかります。実際問題としては依存関係的に明示する必要がないものもあるかもしれませんが、(ロードしておいて文句を言われることとパッケージをロードしていないことに起因するエラーを天秤にかけて)念のためにlibrary関数を用いてロードしています。なお、libraryの代わりにrequire関数を使われる方もいらっしゃいますが、実用上はどちらでも構いません。makeTxDbFromGFF関数でGFF3ファイル名に相当するin_f2を読み込んだ結果をtxdbオブジェクトに格納し、transcripts関数で転写物の座標情報をGRanges形式のhogeオブジェクトに格納しています。最後に「ゲノム配列ファイルに相当するin_f1」と「先ほどのGFF3形式ファイルの中身に相当するhogeオブジェクト」の2つを入力としてgetSeq関数を実行することで、目的のDNAStringSeq形式のfastaオブジェクトを得ることができます。
##から始まっている背景が白くなっている実行結果部分を見ると、確かにfastaオブジェクトはDNAStringSeq形式になっていることがわかります。width列は配列長情報で、たとえば1番最初の転写物の配列長は1086です。この転写物のGRanges形式のstartとendの位置は4769-5854ですので、5,854
- 4769 + 1 =
1,086なので妥当だと判断できます。ここでは表示していませんが、気になるかたはtranscripts(txdb)実行結果に相当するhogeオブジェクトの中身を表示させて確認してみるとよいと思います。
in_f1 <- "Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.dna.toplevel.fa"
in_f2 <- "Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.49.gff3"
library(Rsamtools)
library(GenomicFeatures)
library(Biostrings)
txdb <- makeTxDbFromGFF(in_f2, format="auto")## Error in load_package_gracefully("txdbmaker", "starting with BioC 3.19, ", : Could not load package txdbmaker. Is it installed?
##
## Note that starting with BioC 3.19, calling makeTxDbFromGFF() requires
## the txdbmaker package. Please install it with:
##
## BiocManager::install("txdbmaker")hoge <- transcripts(txdb)## Error in h(simpleError(msg, call)): 引数 'x' の評価中にエラーが起きました (関数 'transcripts' に対するメソッドの選択時): オブジェクト 'txdb' がありませんfasta <- getSeq(FaFile(in_f1), hoge)## Error: 関数 'scanFa' (シグネチャ 'file = "FaFile", param = "DNAStringSet"' に対する) に対する継承メソッドを見付けることができませんfasta## DNAStringSet object of length 1:
## width seq names
## [1] 60 TTGACAATTTCCTGATTCGGACG...ATGAATCCCTCAACTCTATTGCA fig1.8a
以下は、得られた転写物配列情報をFASTA形式ファイルとして保存するところまで含めたスクリプトです。上のスクリプトとの違いは、3行目の出力ファイル名情報を格納するためのout_fオブジェクト作成部分と、最後から2番目の行のwriteXStringSet関数実行部分です。無事実行完了すると、出力ファイル名として指定したファイル(R1.130.1.fasta)が作成されているはずです。ここまでで配列取得の基本形はが完成です。なお、最終行のfasta[1:4]は、「fastaオブジェクト中の最初の4配列分を表示せよ」という意味です。これはhead(fasta, n=4)というコマンドと同じです。headは最初のn個分(デフォルトは6)を出力する関数であるため、n=4として最初の4個分とオプションで指定していることに相当します。
in_f1 <- "Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.dna.toplevel.fa"
in_f2 <- "Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.49.gff3"
out_f <- "R1.130.1.fasta"
library(Rsamtools)
library(GenomicFeatures)
library(Biostrings)
txdb <- makeTxDbFromGFF(in_f2, format="auto")## Error in load_package_gracefully("txdbmaker", "starting with BioC 3.19, ", : Could not load package txdbmaker. Is it installed?
##
## Note that starting with BioC 3.19, calling makeTxDbFromGFF() requires
## the txdbmaker package. Please install it with:
##
## BiocManager::install("txdbmaker")hoge <- transcripts(txdb)## Error in h(simpleError(msg, call)): 引数 'x' の評価中にエラーが起きました (関数 'transcripts' に対するメソッドの選択時): オブジェクト 'txdb' がありませんfasta <- getSeq(FaFile(in_f1), hoge)## Error: 関数 'scanFa' (シグネチャ 'file = "FaFile", param = "DNAStringSet"' に対する) に対する継承メソッドを見付けることができませんwriteXStringSet(fasta, file=out_f, format="fasta", width=50)
fasta[1:4]## Error: subscript contains out-of-bounds indices
さきほどのファイル(R1.130.1.fasta)のdescription行に相当する情報は、上で見えているfastaオブジェクトのnames列です。“ctg01_00128”は、計75配列からなるゲノム配列ファイル(Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.dna.toplevel.fa)の1番最初の配列名です。この情報は、(計3,236配列分あるので画面がざっと流れますが)names(fasta)で一望できます。元のゲノム配列は計75配列(75
contigs)から構成されていますので、種類としては計75種類ということになります。どのコンティグ由来配列が何個あったかは、計3,236要素からなる文字列ベクトルであるnames(fasta)を入力として、以下のようにtable関数を実行すればよいです。実行結果は、たとえば”ctg01_00128”というコンティグ由来転写物は38個、“ctg02_00001”由来の転写物は17個という風に解釈します。
table(names(fasta))##
## fig1.8a
## 1R1.140 1.10.2項のGFF3(配列取得2)
配列取得1では、転写物配列取得の基本形を示しました。以下は、先ほどの配列取得1の出力ファイル(R1.130.1.fasta)を得るスクリプトの再掲です。hogeはtranscripts(txdb)実行結果のGRanges形式のオブジェクト、fastaはhogeの情報を用いて転写物配列を取得したDNAStringSet形式のオブジェクトです。
in_f1 <- "Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.dna.toplevel.fa"
in_f2 <- "Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.49.gff3"
out_f <- "R1.130.1.fasta"
library(Rsamtools)
library(GenomicFeatures)
library(Biostrings)
txdb <- makeTxDbFromGFF(in_f2, format="auto")## Error in load_package_gracefully("txdbmaker", "starting with BioC 3.19, ", : Could not load package txdbmaker. Is it installed?
##
## Note that starting with BioC 3.19, calling makeTxDbFromGFF() requires
## the txdbmaker package. Please install it with:
##
## BiocManager::install("txdbmaker")hoge <- transcripts(txdb)## Error in h(simpleError(msg, call)): 引数 'x' の評価中にエラーが起きました (関数 'transcripts' に対するメソッドの選択時): オブジェクト 'txdb' がありませんfasta <- getSeq(FaFile(in_f1), hoge)## Error: 関数 'scanFa' (シグネチャ 'file = "FaFile", param = "DNAStringSet"' に対する) に対する継承メソッドを見付けることができませんwriteXStringSet(fasta, file=out_f, format="fasta", width=50)
以下は、hogeオブジェクトの最初の3行分を表示させた結果です。最初の2列(seqnamesとranges列)は、その列名を関数として情報抽出に利用できます。
hoge[1:3]## DNAStringSet object of length 3:
## width seq
## [1] 3 TTG
## [2] 15 AATTTCCTGATTCGG
## [3] 5 GTCCC
以下は、fastaオブジェクトの最初の3行分を表示させた結果です。names列の情報に相当するnames(fasta)は、hogeオブジェクトのseqnames列の情報と同じであることがわかります。
fasta[1:3]## Error: subscript contains out-of-bounds indices
以下は、seqnames(hoge)の実行結果です。見慣れないと面食らってしまうかもしれませんが、先程のtable(names(fasta))実行結果と見比べれば、「ctg01_00128が38個ある」や「ctg02_00001が17個ある」といったことがわかります。ここでは示しませんが、table(names(fasta))の実行結果は、table(seqnames(hoge))の実行結果と同じです。また、names(fasta)と同じ見栄えの情報は、as.character(seqnames(hoge))で得られます。さらに、table(seqnames(hoge))と同じ結果は、table(as.character(seqnames(hoge)))でも得られます。いきなりtable(as.character(seqnames(hoge)))を見ると難解だと思うかもしれませんが、このように様々な情報と照らし合わせて、要素ごとに分解して眺めると理解しやすいのではと思います。
seqnames(hoge)## Error: 関数 'seqnames' (シグネチャ 'x = "DNAStringSet"' に対する) に対する継承メソッドを見付けることができません
次は、ranges(hoge)の実行結果です。確かにhogeオブジェクト中のranges列で見えている情報が含まれています。start列とend列に加えてwidth列が見えていますが、これは(end
- start +1)で算出される配列長情報です。
ranges(hoge)## Error: 関数 'ranges' (シグネチャ 'x = "DNAStringSet"' に対する) に対する継承メソッドを見付けることができません
ranges(hoge)の実行結果で見えている情報は、その列名を関数として当該列情報を抽出できます。具体的には、start列の情報はstart(ranges(hoge))、end列の情報はend(ranges(hoge))、そしてwidth列の情報はwidth(ranges(hoge))で取得可能です。以下では、(これをそのまま実行してしまうと、それぞれについて計3,236配列分の要素の情報が一気に表示されて見づらいため)さらにhead関数をつけて最初の6個分に限定して示します。
head(start(ranges(hoge)))## Error in h(simpleError(msg, call)): 引数 'x' の評価中にエラーが起きました (関数 'head' に対するメソッドの選択時): 引数 'x' の評価中にエラーが起きました (関数 'start' に対するメソッドの選択時): 関数 'ranges' (シグネチャ 'x = "DNAStringSet"' に対する) に対する継承メソッドを見付けることができませんhead(end(ranges(hoge)))## Error in h(simpleError(msg, call)): 引数 'x' の評価中にエラーが起きました (関数 'head' に対するメソッドの選択時): 引数 'x' の評価中にエラーが起きました (関数 'end' に対するメソッドの選択時): 関数 'ranges' (シグネチャ 'x = "DNAStringSet"' に対する) に対する継承メソッドを見付けることができませんhead(width(ranges(hoge)))## Error in h(simpleError(msg, call)): 引数 'x' の評価中にエラーが起きました (関数 'head' に対するメソッドの選択時): 引数 'x' の評価中にエラーが起きました (関数 'width' に対するメソッドの選択時): 関数 'ranges' (シグネチャ 'x = "DNAStringSet"' に対する) に対する継承メソッドを見付けることができません
ここで、再度転写物配列情報であるfastaオブジェクトの最初の5行分を表示させます。names列の情報に相当するnames(fasta)はR1.130.1.fastaのdescription行に相当しますが、現状では由来配列名しか示されていないことがわかります。
fasta[1:5]## Error: subscript contains out-of-bounds indices
そこで、これまで示してきたテクニック+αで、names(fasta)の情報を「由来配列名」から「由来配列名_start_end」に変更し、転写物配列がどの配列のどの領域由来かがdescription情報をみればわかるようにします。まず、新たな関数としてpasteを紹介します。pasteは、複数の数値なり文字なりを、任意の文字や記号で連結する関数です。以下に4つ例を示します。最後のコマンドが実際にやろうとしていることに近いものになります。
paste(8, 5, sep="_")## [1] "8_5"paste("an", 5, "pan", sep="__")## [1] "an__5__pan"paste("an", "pon", "tan", sep="")## [1] "anpontan"paste(head(start(ranges(hoge))), head(end(ranges(hoge))), sep="_")## Error in h(simpleError(msg, call)): 引数 'x' の評価中にエラーが起きました (関数 'head' に対するメソッドの選択時): 引数 'x' の評価中にエラーが起きました (関数 'start' に対するメソッドの選択時): 関数 'ranges' (シグネチャ 'x = "DNAStringSet"' に対する) に対する継承メソッドを見付けることができません
ここから本番になっていきますが、まずは変更前のnames(fasta)の最初の6個分の情報を以下に示します。
head(names(fasta))## [1] "fig1.8a"
以下が、names(fasta)の情報を「由来配列名」から「由来配列名_start_end」に変更する本番のコマンドです。あまり横長になっても見づらいので2つの行にわけていますが、paste関数を使って、当該転写物の由来配列名に相当する情報のseqnames(hoge)、当該転写物のstart位置に相当する情報のstart(ranges(hoge))、そして当該転写物のend位置に相当する情報のend(ranges(hoge))を”_“で連結した結果を、names(fasta)に格納(代入)しています。ここでは右辺の情報を統一的にhogeオブジェクトに揃える目的で由来配列名に相当する情報としてseqnames(hoge)を用いましたが、names(fasta)で置き換えてもかまいません。その場合は、右辺のnames(fasta)が「由来配列名」のみに相当し、左辺のnames(fasta)が変更後の「由来配列名_start_end」ということになります。
names(fasta) <- paste(seqnames(hoge), start(ranges(hoge)),
end(ranges(hoge)), sep="_")## Error: 関数 'seqnames' (シグネチャ 'x = "DNAStringSet"' に対する) に対する継承メソッドを見付けることができません
変更後のnames(fasta)の最初の6個分の情報を以下に示すべく、以下で再度head(names(fasta))実行結果を示します。確かに意図通りにstartとendの情報が右側に追加されていることがわかります。
head(names(fasta))## [1] "fig1.8a"
注意点としては、fastaオブジェクトの中身がこの段階で変更されているということです。つまりこの場合は、names列の情報が変更されています。右端が…になっているものがありますが、上のほうで見えている結果を突き合わせることで意図通りに変更されていると確信できると思います。
fasta## DNAStringSet object of length 1:
## width seq names
## [1] 60 TTGACAATTTCCTGATTCGGACG...ATGAATCCCTCAACTCTATTGCA fig1.8a
最後に、names(fasta)の情報を更新したものをファイル(R1.130.2.fasta)に保存するところまでの一連のスクリプトを示します。最終行のfasta[1:3]の実行結果を見れば、確かに出力ファイルのdescription行が正しく変更されていると確信できると思います。
in_f1 <- "Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.dna.toplevel.fa"
in_f2 <- "Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.49.gff3"
out_f <- "R1.130.2.fasta"
library(Rsamtools)
library(GenomicFeatures)
library(Biostrings)
txdb <- makeTxDbFromGFF(in_f2, format="auto")## Error in load_package_gracefully("txdbmaker", "starting with BioC 3.19, ", : Could not load package txdbmaker. Is it installed?
##
## Note that starting with BioC 3.19, calling makeTxDbFromGFF() requires
## the txdbmaker package. Please install it with:
##
## BiocManager::install("txdbmaker")hoge <- transcripts(txdb)## Error in h(simpleError(msg, call)): 引数 'x' の評価中にエラーが起きました (関数 'transcripts' に対するメソッドの選択時): オブジェクト 'txdb' がありませんfasta <- getSeq(FaFile(in_f1), hoge)## Error: 関数 'scanFa' (シグネチャ 'file = "FaFile", param = "DNAStringSet"' に対する) に対する継承メソッドを見付けることができませんnames(fasta) <- paste(seqnames(hoge), start(ranges(hoge)),
end(ranges(hoge)), sep="_")## Error: 関数 'seqnames' (シグネチャ 'x = "DNAStringSet"' に対する) に対する継承メソッドを見付けることができませんwriteXStringSet(fasta, file=out_f, format="fasta", width=50)
fasta[1:3]## Error: subscript contains out-of-bounds indicesR1.150 1.10.3項のリピートマスク
ここでは、リピートマスク前後の乳酸菌ゲノム配列を眺めます。Ensembl (Cunningham et al., Nucleic Acids Res., 2022)のバクテリア版であるEnsemblBacteria (Release 54)上で、乳酸菌の1つであるLactobacillus casei 21/1のページを再訪します。①のDownload DNA sequenceをクリックして、ゲノム配列のダウンロードページに移動します。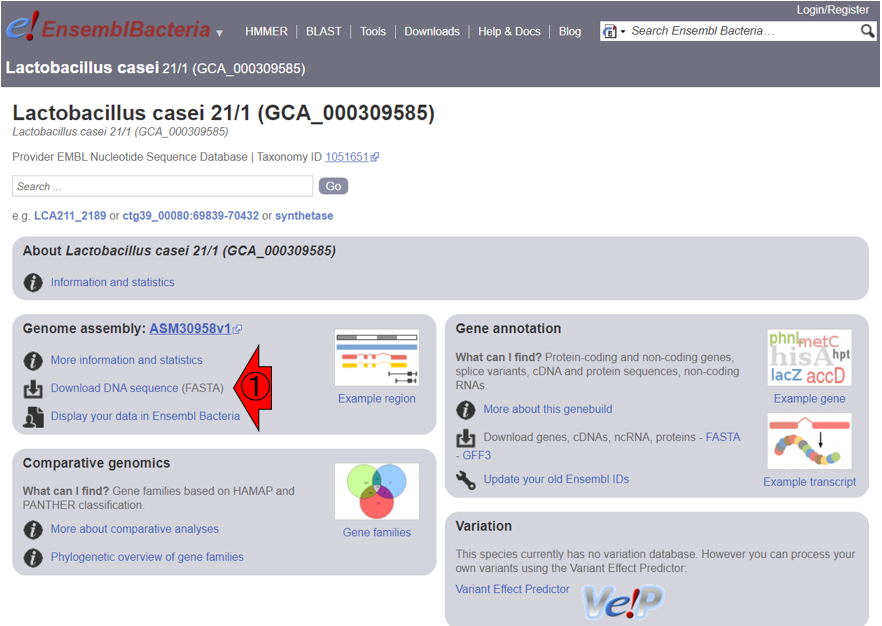
以下がリンク先のスクショです。一番上のCHECKSUMは、ダウンロードしたファイルが壊れていないか(提供元と同一か)を調べるための情報です。一番下のREADMEは、計6種類見えているゲノム配列ファイルについての解説が書かれています。
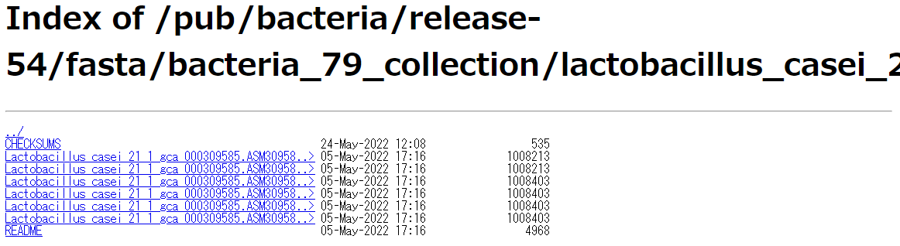
上記の情報はEnsemblBacteria (Release 54)のものですが、リリースの違いによってリンク切れになったりします。それゆえ、以降の解析が数年後でも可能となるよう、念のため以下でもファイルへのリンクを貼っておきます。“Lactobacillus_casei_21_1_gca_000309585.ASM30958v1”までが共通で、それ以降の文字列が異なっていることがわかります。たとえば1-2番目のファイルはは”dna”、3-4番目のファイルは”dna_rm”、そして5-6番目のファイルは”dna_sm”となっていることがわかります。“dna”はマスクされていない配列(unmasked)のことです。“dna_rm”は、RepeatMaskerで同定された反復配列をマスクした(masked)配列のことで、具体的には”N”で置換した配列のことです。“dna_sm”は、RepeatMaskerで同定された反復配列をソフトにマスクした(soft-masked)配列のことです。その次のnonchromosomalは、当該ゲノムの配列決定レベル(解像度)が最高の染色体レベルになったとしても、どの染色体にも割り当てられない断片配列が存在しますので、それらの配列も含めたものです。toplevelは、当該ゲノムの最高の配列決定レベルで得られた配列から構成されるファイルです。この場合は、配列決定精度がコンティグレベル(contig; ctg)ですので、nonchromosomalとtoplevelの中身は同じになります。直感的には、最も”N”が少ないのが”dna”、最も多いのが”dna_rm”です。また、最も”ACGTN”以外の文字が多いのが”dna_sm”ということになります。
- Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.dna.nonchromosomal.fa.gz
- Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.dna.toplevel.fa.gz
- Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.dna_rm.nonchromosomal.fa.gz
- Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.dna_rm.toplevel.fa.gz
- Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.dna_sm.nonchromosomal.fa.gz
- Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.dna_sm.toplevel.fa.gz
soft-maskedがわかりづらいかもしれませんが、これは塩基配列の表記法(nomenclature)はA, C, G, T, Nの5種類だけではないという事実を正しく理解するところからスタートせねばなりません。たとえばプリン塩基(A or G)であることが確かなら、1文字表記のRとします。同様に、ピリミジン塩基(C or T)であることが確かならYです。他にもありますので、全体像を以下にまとめておきます。なお、元情報は1984年に勧告された命名法の論文(Cornish-Bowden A., Nucleic Acids Res., 1985)中のTable 1です。
| 塩基 | 1文字表記 | 表記の由来 |
|---|---|---|
| A or C | M | aMino |
| A or G | R | puRine |
| A or T | W | Weak interaction (2 H bonds) |
| C or G | S | Strong interaction (3 H bonds) |
| C or T | Y | pYrimidine |
| G or T | K | Ketone |
| A or C or G | V | not-T (not-U), V follows U in the alphabet |
| A or C or T | H | not-G, H follows G in the alphabet |
| A or G or T | D | not-C, D follows C in the alphabet |
| C or G or T | B | not-A, B follows A in the alphabet |
| A or C or G or T | N | aNy |
先程までGFF3ファイルであるLactobacillus_casei_21_1_gca_000309585.ASM30958v1.49.gff3とともに読み込んで、転写物配列ファイル(R1.130.2.fasta)作成の元データとして用いたのは、Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.dna.toplevel.faです。以下ではまず、Biostringsパッケージが提供するreadDNAStringSet関数がgzip圧縮ファイルでも読み込めることを示します。DNAStringSeq形式のfastaオブジェクトのwidth列とnames列を見比べることで、width列の配列長で降順(多–>少)にコンティグ(contig;
ctg)が並べられていることがわかります。
in_f <- "Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.dna.toplevel.fa.gz"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta## DNAStringSet object of length 75:
## width seq names
## [1] 362642 GAGGACGCGCACGTGGGCCGTG...CGGTGCTCGCGATATCTGAGTC ctg27_00064 dna:s...
## [2] 208498 AAAACCTGCACATAAAGGACCT...CTAATACCGCATAGATCCAAGA ctg15_00035 dna:s...
## [3] 157092 GTCAAAGGCTGCTGTTTGACCA...CTTGATGAAGCTGAACTGCCCG ctg40_00041 dna:s...
## [4] 145075 AGCTGATGAGCCAGCATTAATG...TATTCGCGTAGTTAGGTGGTCG ctg05_00082 dna:s...
## [5] 137019 TAGAAGCTACTGTATTGGACAG...AAGACCTGTTTTGCTTGGTGGT ctg49_00090 dna:s...
## ... ... ...
## [71] 2084 GAGACGAGGCGATTCGGATTGA...CTCGCGATATCTGAGTCGGAGA ctg68_00104 dna:s...
## [72] 2082 CCCCATTGAGGACGCGCACGTG...TGGTTGCGGCAATTCGTGATAC ctg22_00049 dna:s...
## [73] 1899 TGTTCCTTCTCCCTCGATCCAC...GCATCGTGGAGCTCAATTGAAT ctg62_00065 dna:s...
## [74] 1186 AATCAGTGGTTTTAACACTTTG...CCATCAGTCTTGCAAATCCCTT ctg72_00112 dna:s...
## [75] 981 AAGCTAATCTCTTAAAGCCATT...GAAGAGCCCAAACCGAGAAGCT ctg67_00088 dna:s...
ちなみに、width(fasta)とやることで、配列(この場合はコンティグ)ごとの配列長情報を取得することができます。計75配列あるので、この出力結果は75個の要素からなる数値ベクトルです。1番左側に[1]や[11]や[21]が見えていますが、これは要素番号情報です。たとえば、表示されている数値ベクトルの1番目の要素が362642、11番目の要素が84732、21番目の要素が54844なのだと解釈します。
width(fasta)## [1] 362642 208498 157092 145075 137019 129187 108667 108303 97079 85220
## [11] 84732 82851 77549 73381 71270 63087 62872 61538 60645 55203
## [21] 54844 53115 52123 44573 44411 39905 36629 35796 34344 33427
## [31] 31565 30996 28708 27489 27063 26659 25214 23894 22582 21546
## [41] 20833 19583 15729 15495 15347 14920 14532 14456 13971 12665
## [51] 11741 9875 8984 8827 8373 8327 7299 6625 6268 5549
## [61] 5013 4984 4879 4541 4450 4427 4314 3561 2803 2482
## [71] 2084 2082 1899 1186 981
R1.100
1.10.2項のGFF3(ファイル取得)のところで、「③より、このゲノムサイズは3,215,878
bp」と書いています。この数値は、以下のように計75配列の配列長が格納された数値ベクトルであるwidth(fasta)を入力として、ベクトルの総和を計算するsum関数を実行することで確認できます。
sum(width(fasta))## [1] 3215878
DNAStringSeq形式のfastaオブジェクトを入力としてalphabetFrequency関数を実行すると、fastaオブジェクトの各配列に対して、塩基の種類ごとの出現頻度を返してくれます。ただ、もしalphabetFrequency(fasta)としてしまうと、A,
C, G, T, Nだけでなく、上記の表で示したような「プリン塩基(A or
G)ならR」、「ピリミジン塩基(C or
T)ならY」といったような様々な結果が配列ごとに一気に表示されてしまい見づらくなってしまいます。そのため、以下ではbaseOnly=TRUEというオプションをつけて「A,
C, G, Tとそれ以外」という5種類に要約して表示させています。
alphabetFrequency(fasta, baseOnly=TRUE)## A C G T other
## [1,] 96419 90508 78914 96785 16
## [2,] 55574 45928 51916 55056 24
## [3,] 42001 38922 33789 42380 0
## [4,] 39472 31070 35781 38752 0
## [5,] 37540 32735 29815 36893 36
## [6,] 34247 27717 32885 34337 1
## [7,] 28507 23670 27141 29343 6
## [8,] 28990 23437 26900 28968 8
## [9,] 25883 20792 24303 26101 0
## [10,] 22368 18160 21896 22796 0
## [11,] 22733 20836 18295 22865 3
## [12,] 21759 20859 18548 21682 3
## [13,] 21351 18971 16625 20600 2
## [14,] 20306 15136 17718 20219 2
## [15,] 19105 17766 15239 19158 2
## [16,] 17446 15108 12820 17712 1
## [17,] 16225 15014 14799 16833 1
## [18,] 16366 15551 13489 16132 0
## [19,] 16842 13238 15045 15520 0
## [20,] 15124 11638 13701 14740 0
## [21,] 16161 11516 11904 15231 32
## [22,] 13991 12821 11694 14607 2
## [23,] 13602 12982 11532 14007 0
## [24,] 11933 10183 10502 11955 0
## [25,] 11299 9461 10991 12660 0
## [26,] 10522 10154 8673 10555 1
## [27,] 10198 7654 8813 9964 0
## [28,] 9490 7728 8673 9901 4
## [29,] 9035 7599 8337 9373 0
## [30,] 8910 8529 7174 8813 1
## [31,] 9705 6549 7610 7701 0
## [32,] 7701 7901 6785 8599 10
## [33,] 7323 6110 7427 7848 0
## [34,] 7266 6970 6098 7155 0
## [35,] 7413 6668 5876 7104 2
## [36,] 6917 6590 6032 7120 0
## [37,] 7460 5231 5738 6768 17
## [38,] 7083 5545 4640 6626 0
## [39,] 6424 4932 4768 6440 18
## [40,] 5332 5061 4639 6511 3
## [41,] 4863 5826 5688 4446 10
## [42,] 5925 3718 4271 5664 5
## [43,] 4413 3538 3821 3956 1
## [44,] 4098 3884 3499 4012 2
## [45,] 3946 3203 3812 4386 0
## [46,] 4038 3874 3265 3743 0
## [47,] 4032 3341 3726 3433 0
## [48,] 3903 3348 3456 3748 1
## [49,] 3818 3481 2983 3687 2
## [50,] 3602 2627 2748 3684 4
## [51,] 3686 2201 2500 3352 2
## [52,] 2932 1720 1993 3230 0
## [53,] 2130 2294 2532 2024 4
## [54,] 2688 1769 1760 2604 6
## [55,] 2478 1751 1668 2476 0
## [56,] 2387 2020 1703 2217 0
## [57,] 2107 1563 1651 1978 0
## [58,] 1856 1510 1373 1874 12
## [59,] 1667 1339 1426 1836 0
## [60,] 1686 1047 1131 1685 0
## [61,] 1325 1191 1211 1281 5
## [62,] 1513 963 1126 1380 2
## [63,] 1379 996 1127 1377 0
## [64,] 1385 970 875 1306 5
## [65,] 1165 893 1120 1271 1
## [66,] 1192 991 977 1266 1
## [67,] 1139 969 964 1241 1
## [68,] 1045 831 644 1041 0
## [69,] 792 530 554 927 0
## [70,] 774 578 419 711 0
## [71,] 630 501 483 470 0
## [72,] 551 467 415 649 0
## [73,] 608 314 393 584 0
## [74,] 325 261 280 320 0
## [75,] 268 226 264 223 0
なお、baseOnly=TRUEオプションをつけずにalphabetFrequency(fasta)を実行した結果を、そのままcolSumsの入力として与えることで、入力配列全体のプリン塩基やピリミジン塩基の数がわかります。
in_f <- "Lactobacillus_casei_21_1_gca_000309585.ASM30958v1.dna.toplevel.fa.gz"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
colSums(alphabetFrequency(fasta))## A C G T M R W S Y K V
## 866369 741975 743383 863892 21 88 18 18 89 17 0
## H D B N - + .
## 0 0 0 8 0 0 0R関連情報(第2章)
R2.010 2.1.3項の組合せ計算
page041の1行目で計算している171通りというのは、choose関数で実行できます。
choose(19, 17)## [1] 171R2.020 2.2.2項の図2.4
page047の図2.4aの再現を行います。まずは「(a)実データ」です。-log(p-value)の値が若干異なります(教科書では0.9248で、以下では0.9249234になっていると思います)。理由は不明ですが(単なる転記ミス?!)、まあ誤差範囲といえるでしょう。
data <- matrix(c(222, 86, 149, 228, 105, 124), nrow=3)
data## [,1] [,2]
## [1,] 222 228
## [2,] 86 105
## [3,] 149 124chisq.test(data)$p.value## [1] 0.1188712-log10(chisq.test(data)$p.value)## [1] 0.9249234
次は「(a)仮想データ1」です。
data <- matrix(c(222, 106, 129, 228, 105, 124), nrow=3)
data## [,1] [,2]
## [1,] 222 228
## [2,] 106 105
## [3,] 129 124chisq.test(data)$p.value## [1] 0.9123088-log10(chisq.test(data)$p.value)## [1] 0.03985815
次は「(a)仮想データ2」です。
data <- matrix(c(202, 86, 169, 228, 105, 124), nrow=3)
data## [,1] [,2]
## [1,] 202 228
## [2,] 86 105
## [3,] 169 124chisq.test(data)$p.value## [1] 0.005590402-log10(chisq.test(data)$p.value)## [1] 2.252557
次は、表の中にはありませんが、本文中で述べている「実データを基本として全部10倍の罹患群4,570人
vs. 対照群4,570人」です。
data <- matrix(c(2220, 860, 1490, 2280, 1050, 1240), nrow=3)
data## [,1] [,2]
## [1,] 2220 2280
## [2,] 860 1050
## [3,] 1490 1240chisq.test(data)$p.value## [1] 5.633341e-10-log10(chisq.test(data)$p.value)## [1] 9.249234
最後に「(b)」です。
data <- matrix(c(287, 149, 21, 292, 150, 15), nrow=3)
data## [,1] [,2]
## [1,] 287 292
## [2,] 149 150
## [3,] 21 15chisq.test(data)$p.value## [1] 0.5925849-log10(chisq.test(data)$p.value)## [1] 0.2272494R2.030 2.4.3項の表2.1(準備)
page055の表2.1は、Bioconductorから提供されているBSgenome.Hsapiens.UCSC.hg19(ヒトゲノム配列のパッケージ)とTxDb.Hsapiens.UCSC.hg19.knownGene(上記ヒトゲノム配列パッケージに対応したアノテーション情報のパッケージ)を利用して作成しています。ここではまず、これら2つのパッケージをインストールします。圧縮して提供されているため、ヒトゲノムの一般的なイメージである3GBまではないにせよ、700MB程度はあります。したがって、インストールにはそれなりの時間がかかります(光のWifi環境で約5分とか…)。
BiocManager::install("BSgenome.Hsapiens.UCSC.hg19", update=F)
BiocManager::install("TxDb.Hsapiens.UCSC.hg19.knownGene", update=F)R2.040 2.4.3項の表2.1a
ここではまず、表2.1aの1番左側の列で見えている2-merの並び情報をnarabiオブジェクトとして定義し、必要なパッケージをロードします。
narabi <- c("AA", "AT", "TA", "TT", "CC", "CG", "GC", "GG",
"AC", "AG", "CA", "CT", "GA", "GT", "TC", "TG")
library(Biostrings) #パッケージのロード
library(BSgenome.Hsapiens.UCSC.hg19)#パッケージのロード
以下は、さきほどロードしたBSgenome.Hsapiens.UCSC.hg19と同名のオブジェクトをgenomeという名前に変更したのち、getSeq関数でDNAStringSet object形式のオブジェクトとして抽出した結果をfastaに保存するまでのスクリプトです。最後にfastaオブジェクトの配列名情報を与えるべく、seqnames(genome)で代入しています。
#前処理(指定したパッケージ中のオブジェクト名をgenomeに統一)
genome <- BSgenome.Hsapiens.UCSC.hg19 #renameしているだけです
fasta <- getSeq(genome) #ゲノム塩基配列情報を抽出した結果をfastaに格納
names(fasta) <- seqnames(genome) #description情報を追加している
fasta #確認してるだけです## DNAStringSet object of length 298:
## width seq names
## [1] 249250621 NNNNNNNNNNNNNNNNNNNN...NNNNNNNNNNNNNNNNNNNN chr1
## [2] 243199373 NNNNNNNNNNNNNNNNNNNN...NNNNNNNNNNNNNNNNNNNN chr2
## [3] 198022430 NNNNNNNNNNNNNNNNNNNN...NNNNNNNNNNNNNNNNNNNN chr3
## [4] 191154276 NNNNNNNNNNNNNNNNNNNN...NNNNNNNNNNNNNNNNNNNN chr4
## [5] 180915260 NNNNNNNNNNNNNNNNNNNN...NNNNNNNNNNNNNNNNNNNN chr5
## ... ... ...
## [294] 74652 TTGTAAATTTGAGATACAAA...TACATATTTGAAGATATGTA chr21_gl383580_alt
## [295] 116690 CACTTGGCTTCACGTCTTGA...TAATAAGGTTTACTATCAGT chr21_gl383581_alt
## [296] 162811 TGGCTACATGCGCCTGGTGG...CAGGTTTAAGCGATTCTCCC chr22_gl383582_alt
## [297] 96924 AAAGGAGCCTTCTGGTGGGT...GCCTCGGCCCGGCCGAAGTA chr22_gl383583_alt
## [298] 74013 TGGCTACATGCGCCTGGTGG...AGACCACGCACCCACCAGAA chr22_kb663609_alt
以下は、表2.1の2列目の情報を得る部分のスクリプトです。まず、fastaを入力としてalphabetFrequency関数を実行し、配列ごとのACGT各塩基の出現頻度をカウントした結果をhogeに格納しています。hoge自体は、298行×18列からなる数値行列です。1行目がchr1、2行目がchr2といった感じで、配列ごとの結果が各行に格納されています。各列にはA,
C, G, T,
…の並びで塩基ごとのカウント結果が格納されています。それゆえ、colSums関数を用いて列ごとの総和を計算してやれば、fastaオブジェクト全体のAの数、Cの数、Gの数、Tの数の情報が得られます。実行結果であるfrequencyオブジェクトはベクトルであり、1番目の要素がAの数、2番目の要素がCの数、といった感じで格納されています。それゆえ、sum(frequency[2:3])/sum(frequency[1:4])のような計算をしてあげれば、GC含量の情報(=
0.4101656)が得られるのです。expectedオブジェクトを得る部分は、はあくまでも理解(説明)のしやすさを優先させています。リアルの研究や投稿論文のSupplementなどで提供する場合は、直前で得たGCcontentオブジェクトの情報を使うなどして一般化したコーディングを行いますし、行ってください。
#本番(expected)
hoge <- alphabetFrequency(fasta) #A,C,G,T,..の数を配列ごとにカウントした結果をhogeに格納
frequency <- colSums(hoge) #列ごとの総和をfrequencyに格納
GCcontent <- sum(frequency[2:3])/sum(frequency[1:4])#GC含量を計算(0.4101656)
expected <- c(rep(0.295*0.295, 4), rep(0.205*0.205, 4), rep(0.205*0.295, 8))#べた書きでnarabiの並びで期待値を算出
expected #確認してるだけです## [1] 0.087025 0.087025 0.087025 0.087025 0.042025 0.042025 0.042025 0.042025
## [9] 0.060475 0.060475 0.060475 0.060475 0.060475 0.060475 0.060475 0.060475
以下は、表2.1の3列目の情報を得る部分のスクリプトです。まず、fastaを入力としてdinucleotideFrequency関数を実行し、配列ごとの2連続塩基の出現頻度をカウントした結果をhogeに格納しています。次に、colSums関数を用いてhogeオブジェクトの列ごとの総和を計算しています。実行結果であるfrequencyオブジェクトはベクトルであり、1番目の要素がAAの数、2番目の要素がACの数、といった感じで格納されています。これは出現頻度情報なので、frequency/sum(frequency)の計算をしてあげることで、出現確率情報に変換することができます。それがprobabilityオブジェクトを得ている部分です。最後にnarabiオブジェクトの並びにしたいので、probability[narabi]を実行して順番を入れ替えています。
#本番(observed)
hoge <- dinucleotideFrequency(fasta, as.prob=F)#連続塩基の出現頻度情報をhogeに格納
frequency <- colSums(hoge) #列ごとの総和をfrequencyに格納
probability <- frequency/sum(frequency)#出現確率を計算した結果をprobabilityに格納
observed <- probability[narabi] #narabiの順番に変更
observed #確認してるだけです## AA AT TA TT CC CG
## 0.097420495 0.076962815 0.065323716 0.097642841 0.052404186 0.009991348
## GC GG AC AG CA CT
## 0.042837423 0.052448019 0.050360014 0.070005838 0.072591985 0.070022548
## GA GT TC TG
## 0.059412950 0.050457091 0.059408565 0.072710165
以下は、表2.1a全体の情報を得るスクリプトです。シンプルに3つのベクトル(expected,
observed, and
observed/expected)をcbind関数を用いて列方向に結合(column
bind)しているだけです。
table2.1a <- cbind(expected, observed, observed/expected)
head(table2.1a) #最初の6行分を表示## expected observed
## AA 0.087025 0.097420495 1.1194541
## AT 0.087025 0.076962815 0.8843759
## TA 0.087025 0.065323716 0.7506316
## TT 0.087025 0.097642841 1.1220091
## CC 0.042025 0.052404186 1.2469765
## CG 0.042025 0.009991348 0.2377477R2.050 2.4.3項の表2.1b
ここではまず、転写開始点(TSS)上流1,000塩基の領域を調べたいので、その数値情報をparam_upstreamオブジェクトに与えています。次に、TxDb.Hsapiens.UCSC.hg19.knownGene(アノテーション情報のパッケージ)をロードして、パッケージ名と同じTxDbという形式のオブジェクトをtxdbという名前に変更(rename)しています。txdbの表示結果の詳細を理解する必要はなく、「こんな感じに見えていたらアノテーション情報がtxdbオブジェクトの中にちゃんと格納されているのだという程度でよい」です。
param_upstream <- 1000 #転写開始点上流の塩基配列数を指定
library(TxDb.Hsapiens.UCSC.hg19.knownGene)#パッケージのロード
txdb <- TxDb.Hsapiens.UCSC.hg19.knownGene #renameしているだけです
txdb #確認してるだけです## TxDb object:
## # Db type: TxDb
## # Supporting package: GenomicFeatures
## # Data source: UCSC
## # Genome: hg19
## # Organism: Homo sapiens
## # Taxonomy ID: 9606
## # UCSC Table: knownGene
## # Resource URL: http://genome.ucsc.edu/
## # Type of Gene ID: Entrez Gene ID
## # Full dataset: yes
## # miRBase build ID: GRCh37
## # transcript_nrow: 82960
## # exon_nrow: 289969
## # cds_nrow: 237533
## # Db created by: GenomicFeatures package from Bioconductor
## # Creation time: 2015-10-07 18:11:28 +0000 (Wed, 07 Oct 2015)
## # GenomicFeatures version at creation time: 1.21.30
## # RSQLite version at creation time: 1.0.0
## # DBSCHEMAVERSION: 1.1
以下は、転写開始点(TSS)上流1,000塩基の配列情報を得る部分のスクリプトです。まず、txdbオブジェクトから遺伝子の座標情報を染色体の座標情報順で取得した結果をgnオブジェクトに保存しています。次に、gnオブジェクトを入力として、5’上流領域(5’
flanking
region)の座標情報を取得するためのflank関数を実行することで、上流1,000塩基分の座標情報を取得しています。そして、上流1,000塩基分の範囲情報からなるhogeオブジェクトを、getSeq関数実行時のオプションとして与えています。fasta実行結果の配列長(width列)が全て1,000塩基であることから、なんとなくうまくとれてるっぽいことがわかります。
#本番
gn <- sort(genes(txdb)) #遺伝子の座標情報を取得
hoge <- flank(gn, width=param_upstream)#指定した範囲の座標情報を取得
fasta <- getSeq(genome, hoge) #指定した範囲の塩基配列情報を取得
fasta #確認してるだけです## DNAStringSet object of length 23056:
## width seq names
## [1] 1000 ACACATGCTACCGCGTCCAGG...CACCATTTTTCTTTTCGTTAA 100287102
## [2] 1000 GCTATTATCACCTATATTTTC...ACGAGTGAAACGAATAACTCT 79501
## [3] 1000 GAATTAGGCTTCTGCTGCCCT...GAGGCAGAGAAAAAGGCGGGG 643837
## [4] 1000 CGGGGAGCCCCGAGGCCCTGG...ACCTCCCCCCAGCTTGGGCCA 148398
## [5] 1000 CGGCGGGGCTCCTATGCAAAG...GCTGCGGGCGGGAGCGGCGGG 339451
## ... ... ...
## [23052] 1000 GGTGAGCCAATCCTGACTCCA...CCGGAAGTGGAAATCTCAGCC 283788
## [23053] 1000 AGCCCTCCACACAAGGGGCTC...CGTTGTTTCTTCCTCTCCAAC 100507412
## [23054] 1000 CGGGGCCCAGGGAGTGGGCGG...CCGGGCAGGCCTCCTGGCTGC 728410
## [23055] 1000 CGGGGCCCAGGGAGTGGGCGG...CCGGGCAGGCCTCCTGGCTGC 100653046
## [23056] 1000 CAGGCTGAGCCCTGCAACGCG...TTCCGGCCGGGGCTCACCGCG 100288687
以下は、表2.1bの1列目の情報を得る部分のスクリプトです。まず、fastaを入力としてalphabetFrequency関数を実行し、配列ごとのACGT各塩基の出現頻度をカウントした結果をhogeに格納しています。hoge自体は、298行×18列からなる数値行列です。1行目がchr1、2行目がchr2といった感じで、配列ごとの結果が各行に格納されています。各列にはA,
C, G, T,
…の並びで塩基ごとのカウント結果が格納されています。それゆえ、colSums関数を用いて列ごとの総和を計算してやれば、つまりfastaオブジェクト全体のAの数、Cの数、Gの数、Tの数の情報が得られます。実行結果であるfrequencyオブジェクトはベクトルであり、1番目の要素がAの数、2番目の要素がCの数、といった感じで格納されています。それゆえ、sum(frequency[2:3])/sum(frequency[1:4])のような計算をしてあげれば、GC含量の情報(=
0.518723)が得られるのです。expectedオブジェクトを得る部分は、はあくまでも理解(説明)のしやすさを優先させています。リアルの研究や投稿論文のSupplementなどで提供する場合は、直前で得たGCcontentオブジェクトの情報を使うなどして一般化したコーディングを行いますし、行ってください。
#本番(expected)
hoge <- alphabetFrequency(fasta) #A,C,G,T,..の数を配列ごとにカウントした結果をhogeに格納
frequency <- colSums(hoge) #列ごとの総和をfrequencyに格納
GCcontent <- sum(frequency[2:3])/sum(frequency[1:4]) #GC含量を計算(0.518723)
expected <- c(rep(0.24*0.24, 4), rep(0.26*0.26, 4), rep(0.26*0.24, 8))#べた書きでnarabiの並びで期待値を算出
expected #確認してるだけです## [1] 0.0576 0.0576 0.0576 0.0576 0.0676 0.0676 0.0676 0.0676 0.0624 0.0624
## [11] 0.0624 0.0624 0.0624 0.0624 0.0624 0.0624
以下は、表2.1bの2列目の情報を得る部分のスクリプトです。まず、fastaを入力としてdinucleotideFrequency関数を実行し、配列ごとの2連続塩基の出現頻度をカウントした結果をhogeに格納しています。次に、colSums関数を用いてhogeオブジェクトの列ごとの総和を計算しています。実行結果であるfrequencyオブジェクトはベクトルであり、1番目の要素がAAの数、2番目の要素がACの数、といった感じで格納されています。これは出現頻度情報なので、frequency/sum(frequency)の計算をしてあげることで、出現確率情報に変換することができます。それがprobabilityオブジェクトを得ている部分です。最後にnarabiオブジェクトの並びにしたいので、probability[narabi]を実行して順番を入れ替えています。
#本番(observed)
hoge <- dinucleotideFrequency(fasta, as.prob=F)#連続塩基の出現頻度情報をhogeに格納
frequency <- colSums(hoge) #列ごとの総和をfrequencyに格納
probability <- frequency / sum(frequency)#出現確率を計算した結果をprobabilityに格納
observed <- probability[narabi] #narabiの順番に変更
observed #確認してるだけです## AA AT TA TT CC CG GC
## 0.07197790 0.04949846 0.04250150 0.06976353 0.08297498 0.03794700 0.06865247
## GG AC AG CA CT GA GT
## 0.08154514 0.04879359 0.07227958 0.06785304 0.07179828 0.06008939 0.04773959
## TC TG
## 0.06034721 0.06623833
以下は、表2.1b全体の情報を得るスクリプトです。シンプルに3つのベクトル(expected,
observed, and
observed/expected)をcbind関数を用いて列方向に結合(column
bind)しているだけです。
table2.1b <- cbind(expected, observed, observed/expected)
head(table2.1b) #最初の6行分を表示## expected observed
## AA 0.0576 0.07197790 1.2496164
## AT 0.0576 0.04949846 0.8593482
## TA 0.0576 0.04250150 0.7378733
## TT 0.0576 0.06976353 1.2111723
## CC 0.0676 0.08297498 1.2274405
## CG 0.0676 0.03794700 0.5613462R2.060 2.4.3項の表2.1(出力)
R2.040とR2.050で得た2つのオブジェクト(table2.1aとtable2.1b)を、narabiの情報を含めてファイル(table2.1.txt)に出力するスクリプトです。
####ファイルに保存
out_f <- "table2.1.txt" #出力ファイル名を指定してout_fに格納
tmp <- cbind(narabi, table2.1a, table2.1b)#保存したい情報をtmpに格納
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存R2.070 2.4.3項の例題2.2
例題2.2の内容も、表示させたい桁数を指定するやり方の提供を兼ねて、念のためここでも示しておきます。Saxonov論文のTable 1より、1 kb upstream resionsのGC含量は0.53、Observed CpG fractionは0.042であることがわかります。まずは「(1)このGC含量(0.53)を用いてCCの期待値を小数第4位まで示せ」を行います。
# CCの期待値
GCcontent <- 0.53 #GC含量
C <- GCcontent/2 #Cの期待値はGC含量の1/2
#結果の基本形
C*C #CCの期待値## [1] 0.070225#表示桁数を指定
param_digit <- 4 #表示させたい桁数
round(C*C, digits=param_digit) #小数点第(param_digit + 1)位で四捨五入した結果## [1] 0.0702次に、「(2) このGC含量(0.53)を用いてGAの期待値を小数第4位まで示せ」を行います。
GCcontent <- 0.53 #GC含量
G <- GCcontent/2 #Gの期待値はGC含量の1/2
A <- (1 - GCcontent)/2 #Aの期待値は(1 - GC含量)の1/2
#結果の基本形
G*A #GAの期待値## [1] 0.062275#表示桁数を指定
param_digit <- 4 #表示させたい桁数
round(G*A, digits=param_digit) #小数点第(param_digit + 1)位で四捨五入した結果## [1] 0.0623次に「(3) 上記1kb upstream regionsの情報(つまりGC含量は0.53、Observed CpG fractionは0.042)を用いてCGの観測値/期待値を少数第3位まで示せ」をやります。
param_digit <- 3 #表示させたい桁数
expected <- C*G #CGの期待値
observed <- 0.042 #問題文から与えられているCGの観測値
round(observed/expected, digits=param_digit)#小数点第(param_digit + 1)位で四捨五入した結果## [1] 0.598R2.080 2.4.4項の性能評価指標
page057の8-9行目で述べている「…混同行列を内部的に作成し、それをもとに上記の感度や特異度などを算出して提示する…」をCRANから提供されているcaretというRパッケージを用いて実践します。caretは、機械学習や回帰系のRパッケージとして有名です。その原著論文(Khun M., J Stat Software, 2008)の引用回数は、7,000弱です(2023年9月18日調べ)。ここではまず、パッケージをインストールします。
BiocManager::install("caret", update=F)以下は本番のコードです。まず、ヒトゲノム上のある範囲に計5つの領域が存在し、それぞれの領域はCpGアイランド(CGI)またはそれ以外(non-CGI)のいずれかのラベルが付与されていると仮定します(これが真実の情報)。この範囲に存在する計5つの領域の塩基配列情報をそれぞれ与えて、CGIかどうかを判定させた結果が手元にあったとします(これが予測の結果)。この要素数が同じ2つのベクトル(truthdayoとpredicted)を入力としてcaretパッケージが提供するconfusionMatrix関数を実行すると、正確度(Accuracy)や感度(Sensitivity)などの結果が得られます。
truthdayo <- c("CGI", "non-CGI", "CGI", "CGI", "non-CGI")#真実の情報
predicted <- c("CGI", "CGI", "CGI", "CGI", "CGI") #予測の結果
library(caret) #パッケージのロード
confusionMatrix(data = factor(predicted), #予測の結果はdataのところ
reference = factor(truthdayo),#真実の情報はreferenceのところ
positive = "CGI") #Positiveを定義## Confusion Matrix and Statistics
##
## Reference
## Prediction CGI non-CGI
## CGI 3 2
## non-CGI 0 0
##
## Accuracy : 0.6
## 95% CI : (0.1466, 0.9473)
## No Information Rate : 0.6
## P-Value [Acc > NIR] : 0.6826
##
## Kappa : 0
##
## Mcnemar's Test P-Value : 0.4795
##
## Sensitivity : 1.0
## Specificity : 0.0
## Pos Pred Value : 0.6
## Neg Pred Value : NaN
## Prevalence : 0.6
## Detection Rate : 0.6
## Detection Prevalence : 1.0
## Balanced Accuracy : 0.5
##
## 'Positive' Class : CGI
## 以下は、よりシンプルに書いたスクリプトです。文字列ベクトルではなく、0
or
1の数値ベクトルを入力として与えています。confusionMatrix関数は”CGI”
or
“non-CGI”のラベル情報をアルファベット順にソートするため、対応するラベルを”0”
or
“1”にしてpositive = "0"をオプションとして与えることで、得られる混同行列の左右の列を意図通りに並べています。
truthdayo <- c(0, 1, 0, 0, 1) #真実の情報
predicted <- c(0, 0, 0, 0, 0) #予測の結果
library(caret) #パッケージのロード
confusionMatrix(data = factor(predicted), #予測の結果はdataのところ
reference = factor(truthdayo),#真実の情報はreferenceのところ
positive = "0") #Positiveを定義## Confusion Matrix and Statistics
##
## Reference
## Prediction 0 1
## 0 3 2
## 1 0 0
##
## Accuracy : 0.6
## 95% CI : (0.1466, 0.9473)
## No Information Rate : 0.6
## P-Value [Acc > NIR] : 0.6826
##
## Kappa : 0
##
## Mcnemar's Test P-Value : 0.4795
##
## Sensitivity : 1.0
## Specificity : 0.0
## Pos Pred Value : 0.6
## Neg Pred Value : NaN
## Prevalence : 0.6
## Detection Rate : 0.6
## Detection Prevalence : 1.0
## Balanced Accuracy : 0.5
##
## 'Positive' Class : 0
## 以下は、感度を下げて特異度を上げるべく、予測結果を変えたスクリプトです。
truthdayo <- c(0, 1, 0, 0, 1) #真実の情報
predicted <- c(1, 1, 1, 0, 1) #予測の結果
library(caret) #パッケージのロード
confusionMatrix(data = factor(predicted), #予測の結果はdataのところ
reference = factor(truthdayo),#真実の情報はreferenceのところ
positive = "0") #Positiveを定義## Confusion Matrix and Statistics
##
## Reference
## Prediction 0 1
## 0 1 0
## 1 2 2
##
## Accuracy : 0.6
## 95% CI : (0.1466, 0.9473)
## No Information Rate : 0.6
## P-Value [Acc > NIR] : 0.6826
##
## Kappa : 0.2857
##
## Mcnemar's Test P-Value : 0.4795
##
## Sensitivity : 0.3333
## Specificity : 1.0000
## Pos Pred Value : 1.0000
## Neg Pred Value : 0.5000
## Prevalence : 0.6000
## Detection Rate : 0.2000
## Detection Prevalence : 0.2000
## Balanced Accuracy : 0.6667
##
## 'Positive' Class : 0
##